|
description
|
file link
|
|
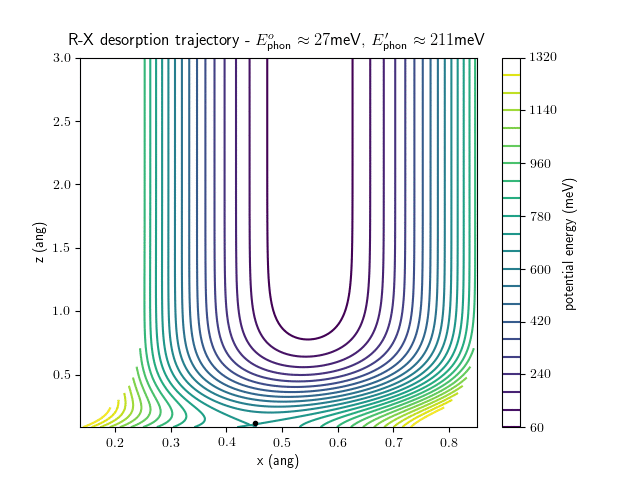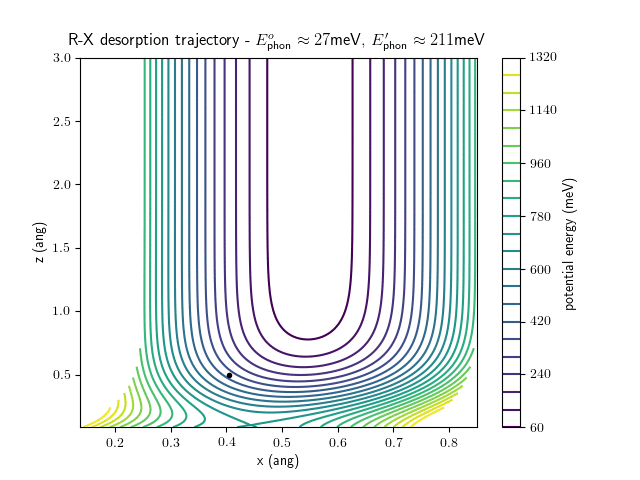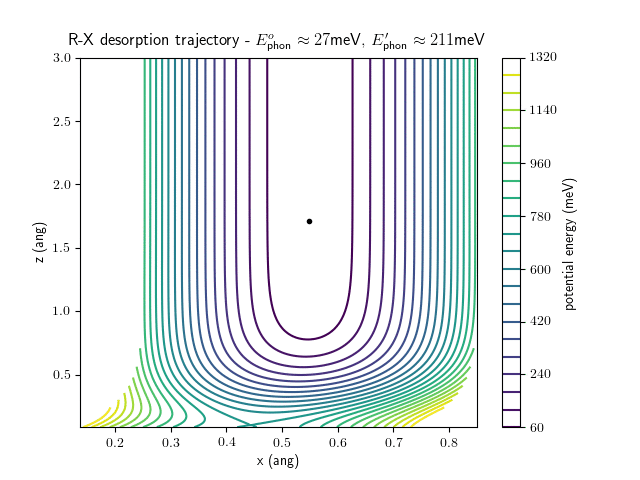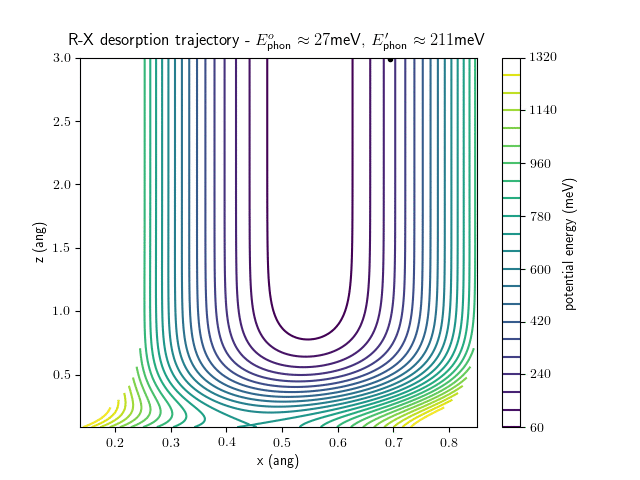
desorption
|
download
|
|
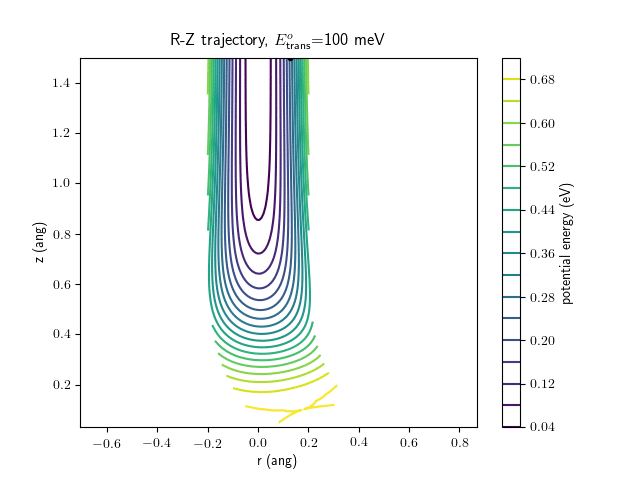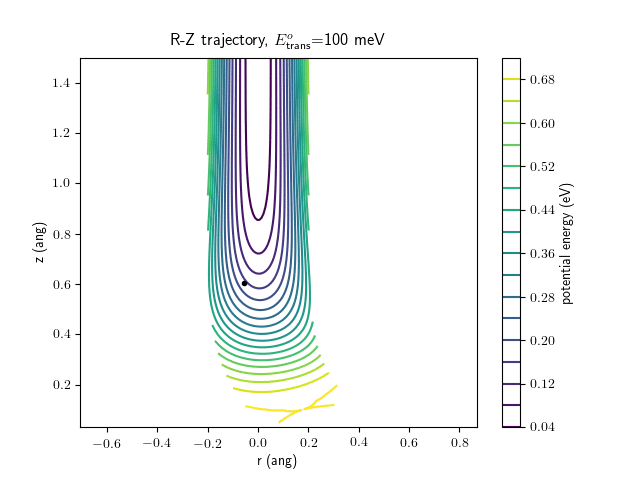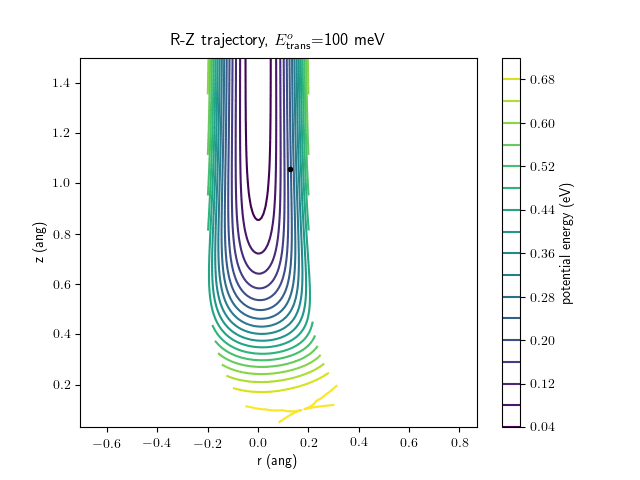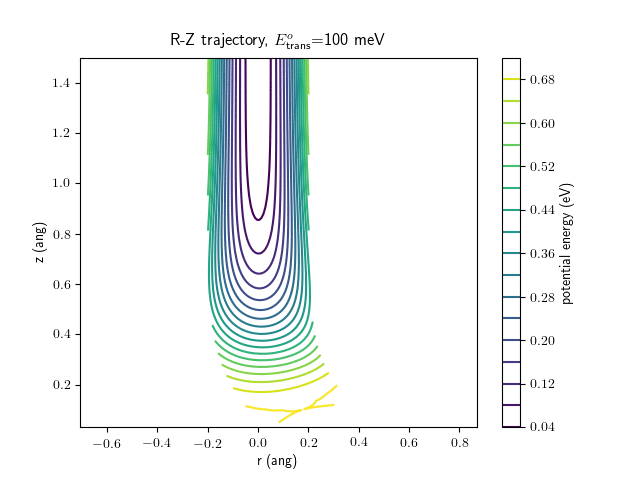
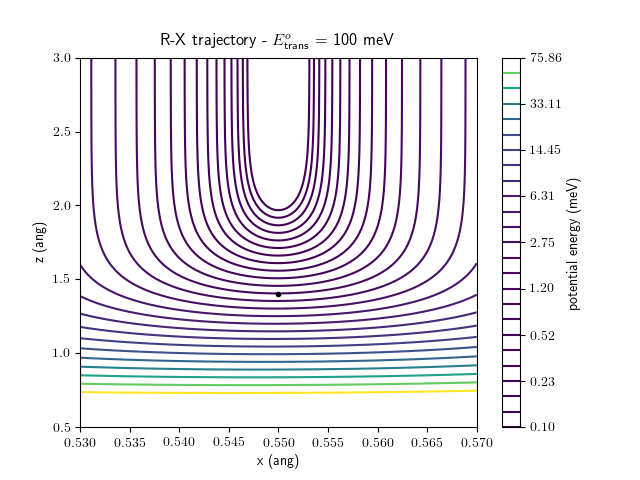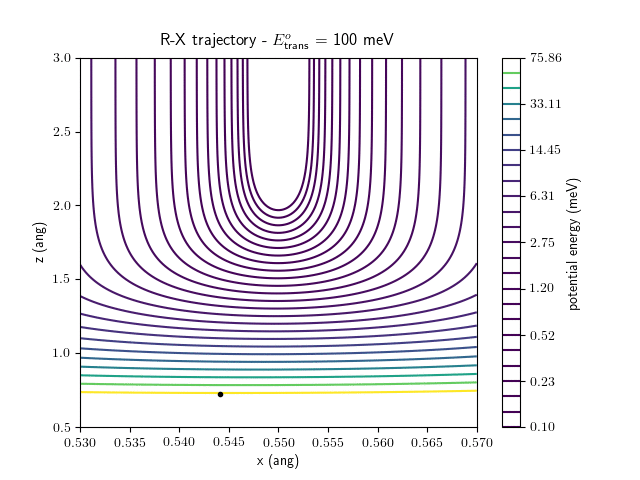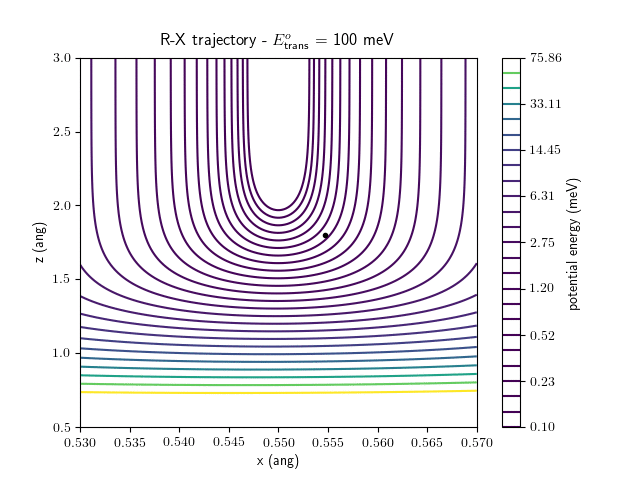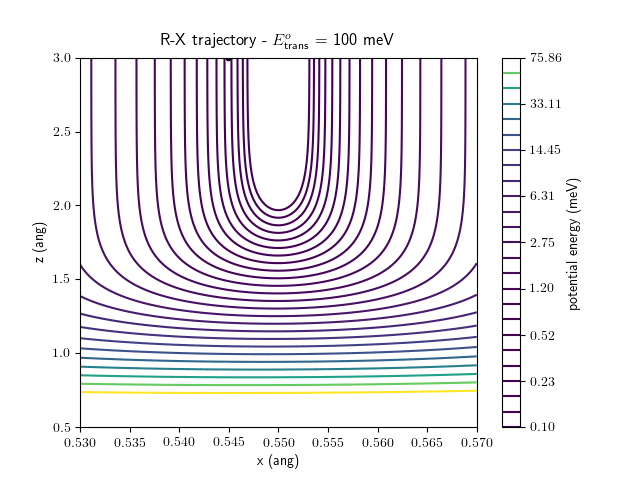
Etrans = 100 meV, Evib = 0 meV
|
download
|
|
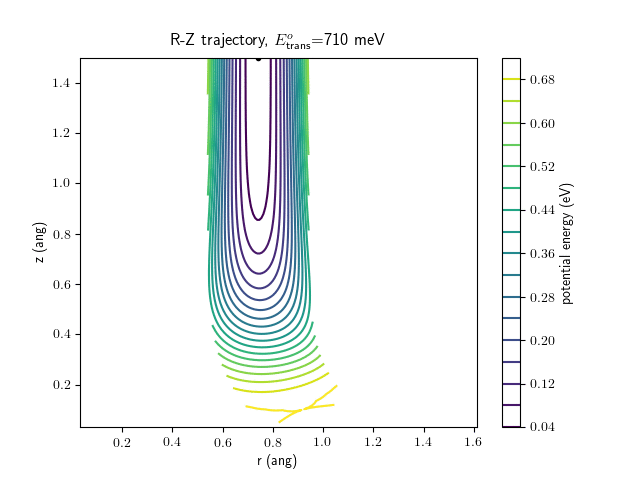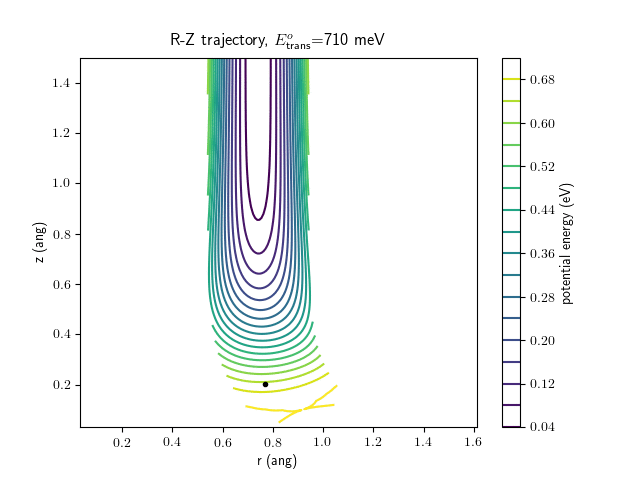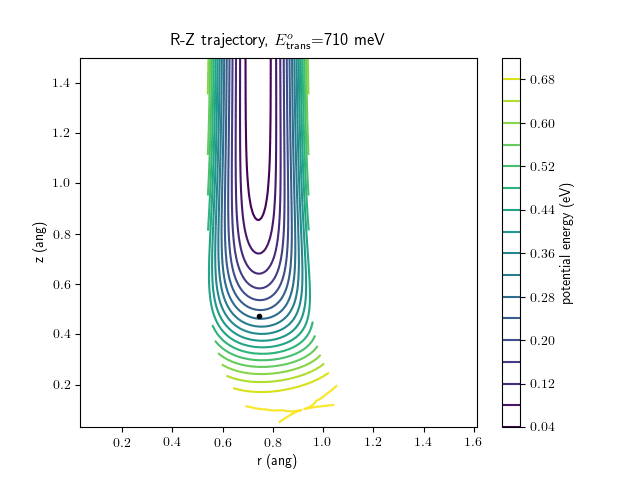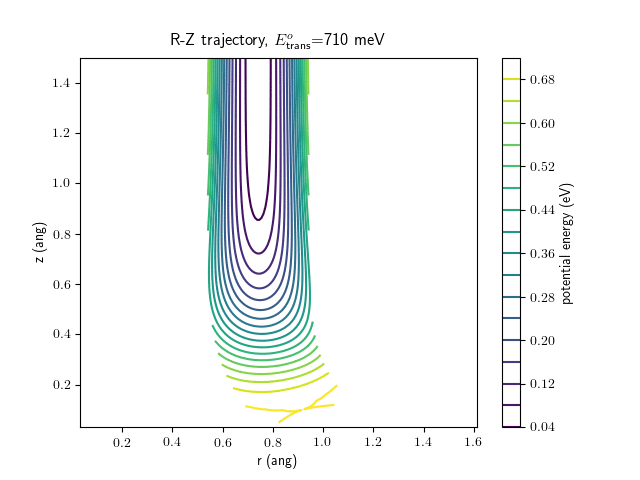
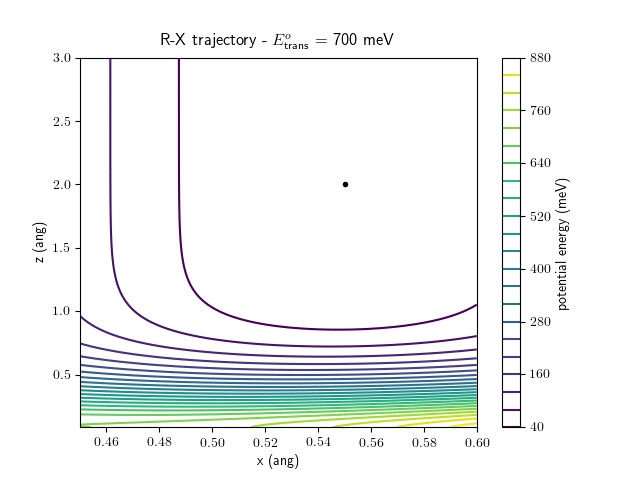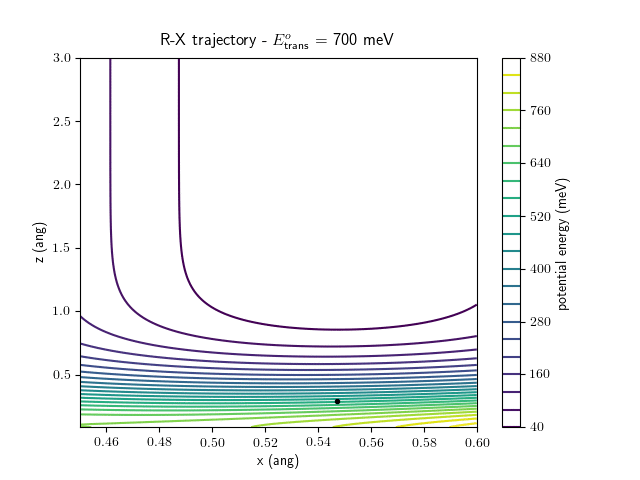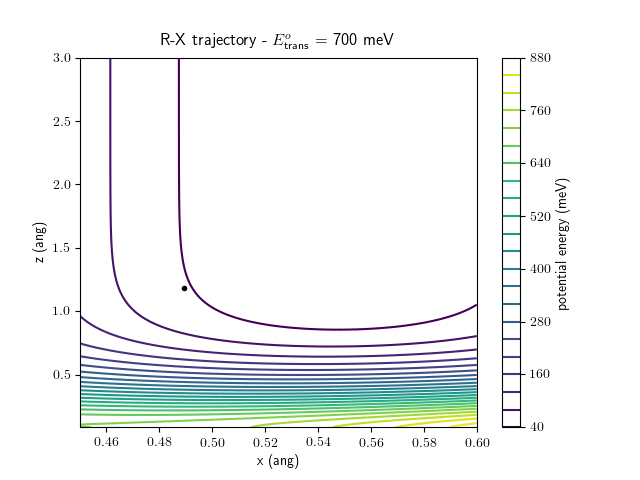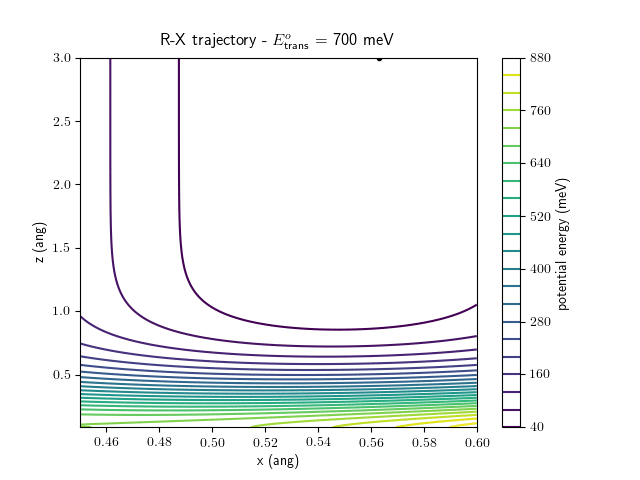
Etrans = 710 meV, Evib = 0 meV
|
download
|
|
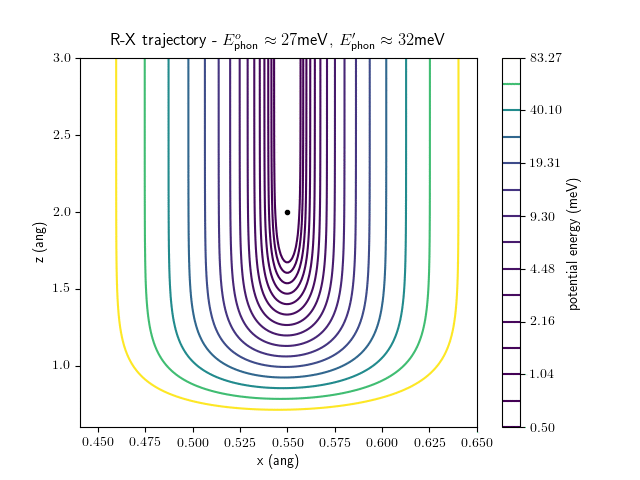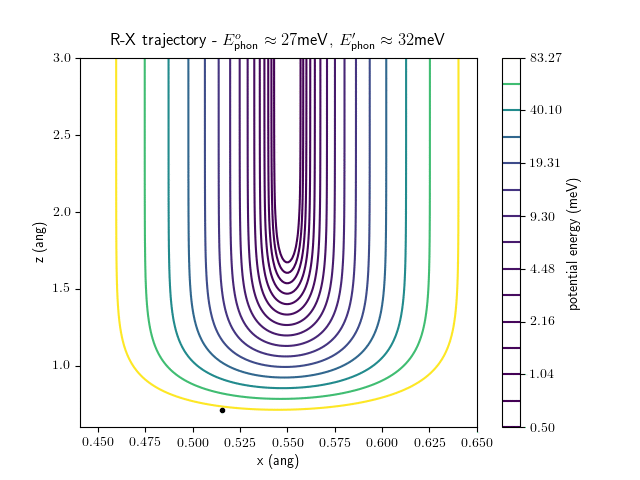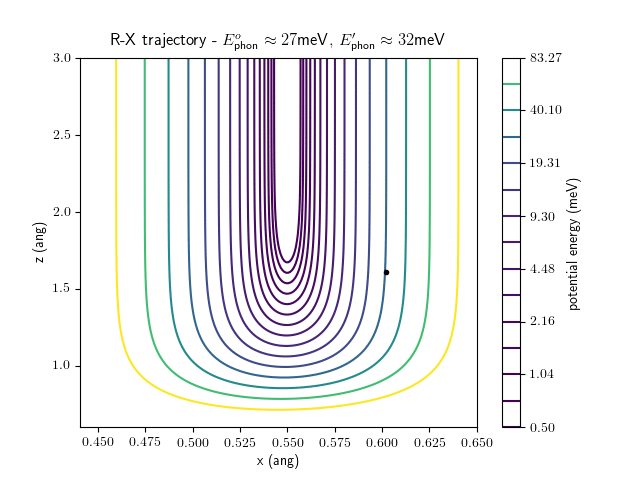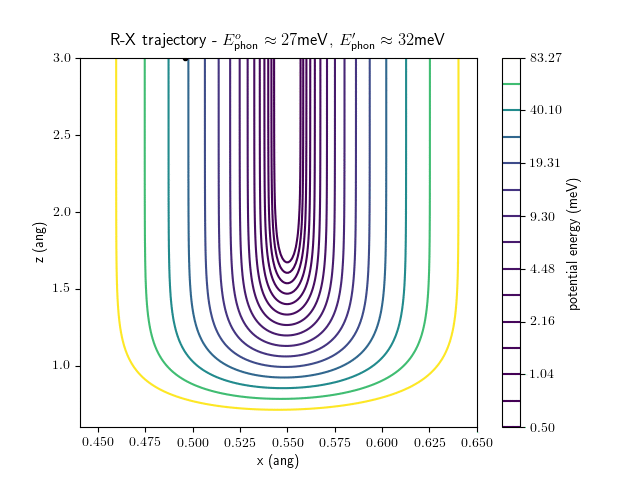
Etrans = 100 meV, Evib = $\frac{1}{2}\hbar\omega$
|
download
|
|
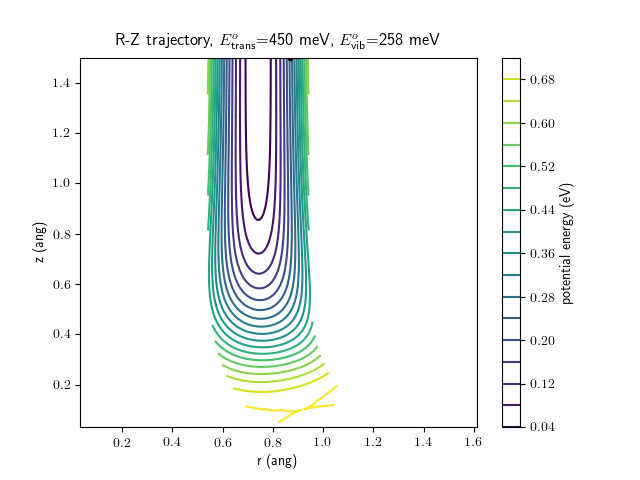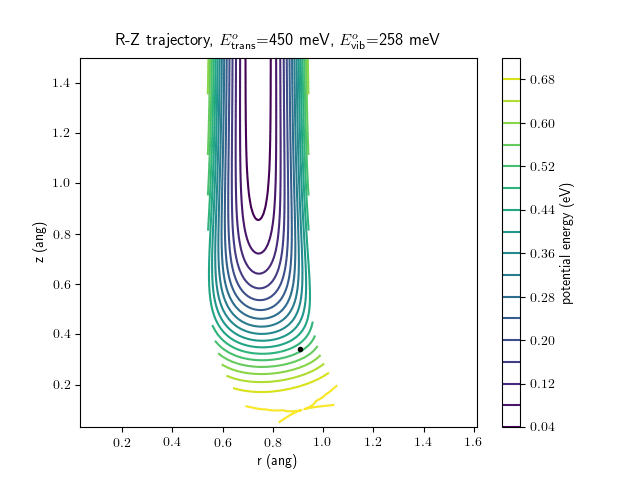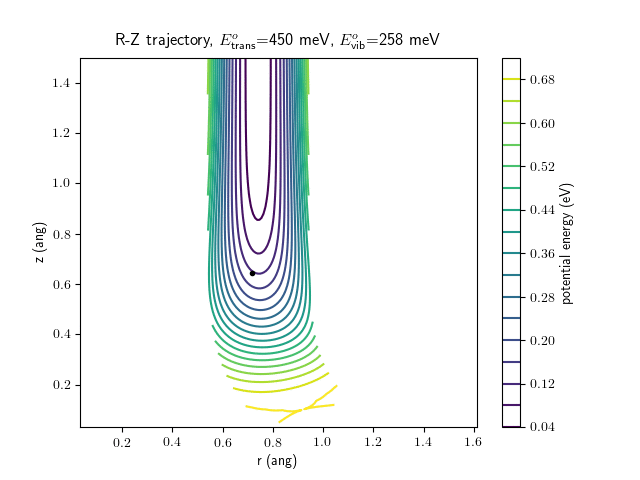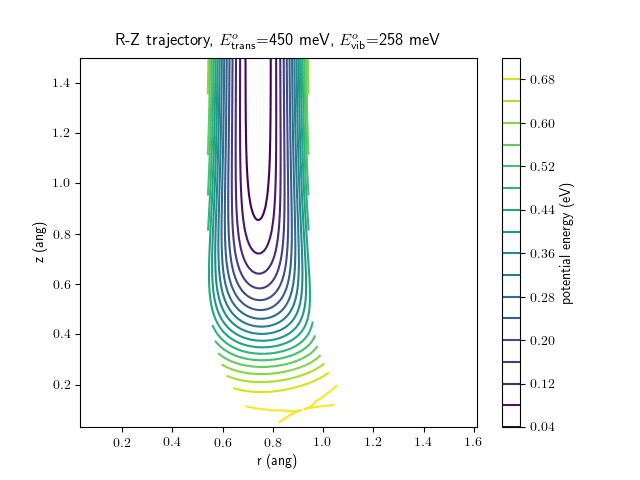
Etrans = 450 meV, Evib = $\frac{1}{2}\hbar\omega$
|
download
|
The trajectory animations demonstrate that the program appears to be working correctly and also show that at our beam energies ($\approx$100meV) the translational motion is almost completely decoupled from the vibrational.
The animations at higher beam energies, however, show appreciable V-T coupling, as does the animation for the desorbing molecule.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}